

Comment le coronavirus infecte les cellules – et pourquoi le variant Delta est si dangereux

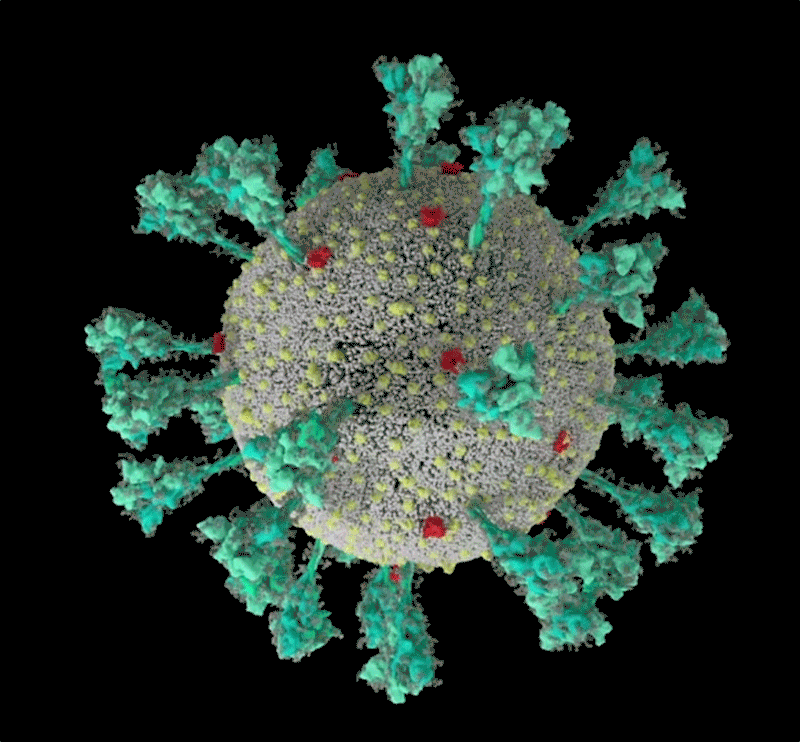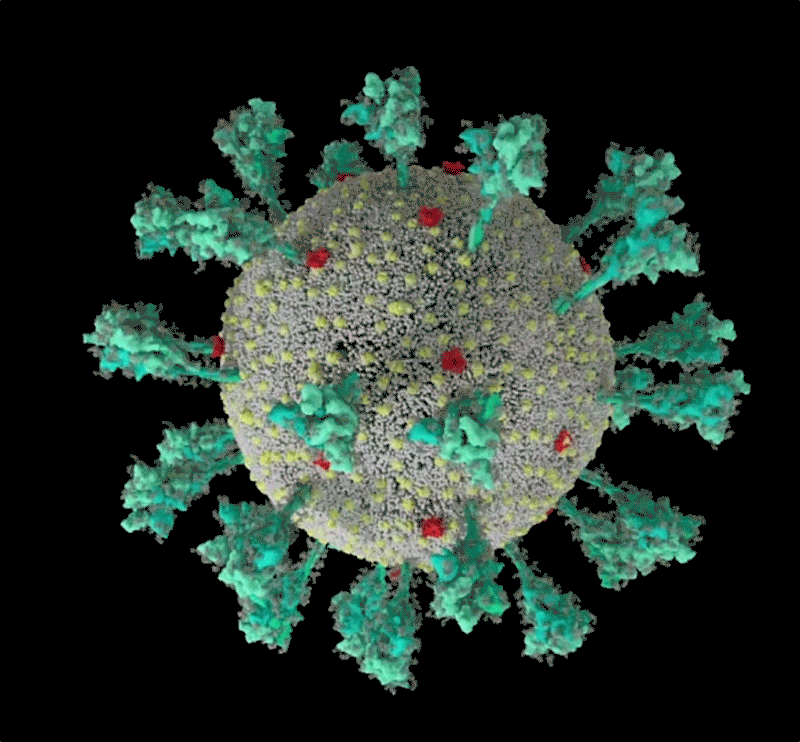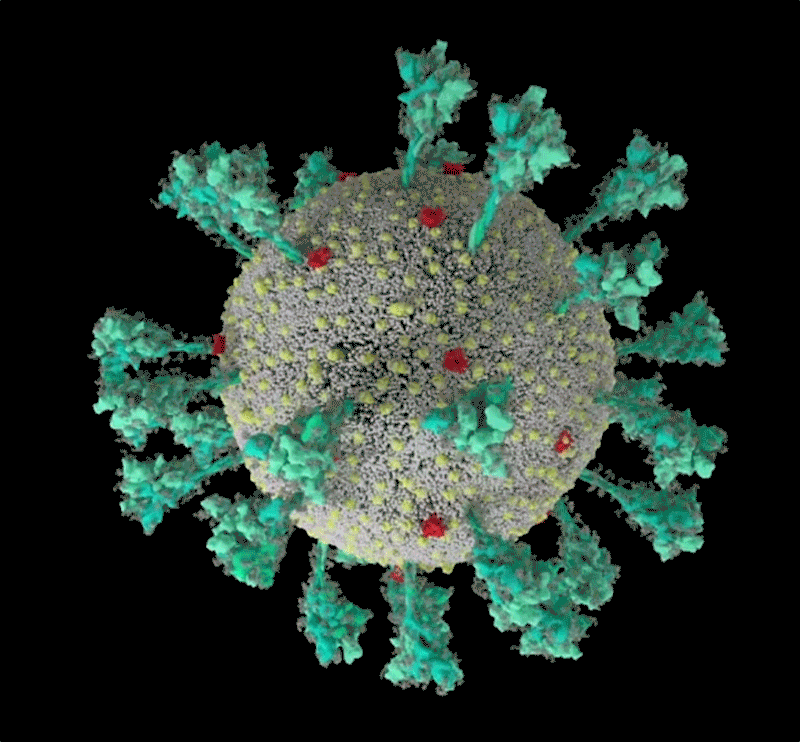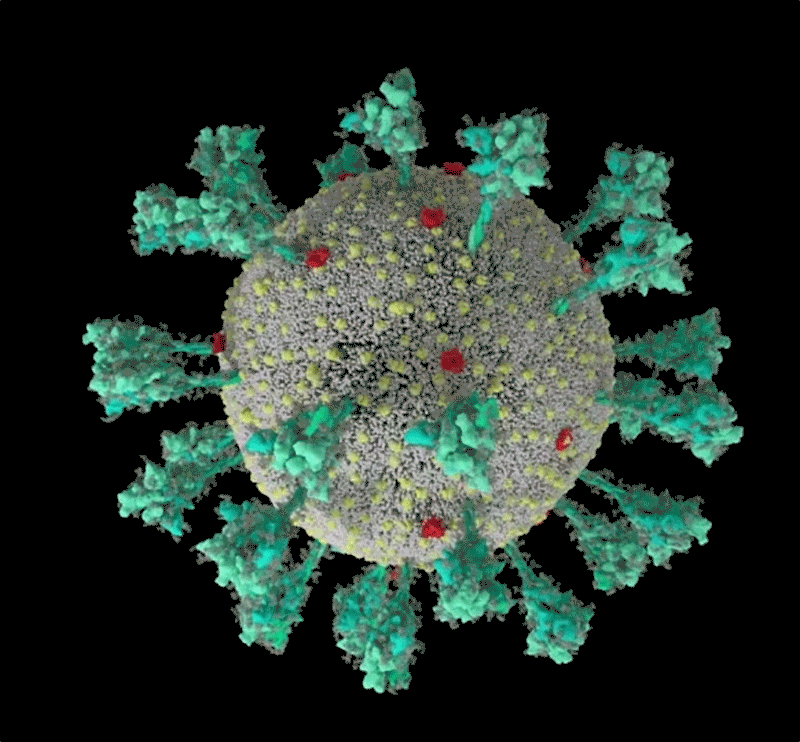
Une simulation informatique de la structure du coronavirus SARS-CoV-2. Crédit : Janet Iwasa, Université de l’Utah
L’art du camouflage
Le coronavirus arbore un luxueux manteau de sucre. « C’est frappant », a pensé Rommie Amaro, regardant sa simulation informatique de l’une des protéines de pointe de marque du SRAS-CoV-2, qui dépasse de la surface du virus. Il était enveloppé de molécules de sucre, appelées glycanes. « Quand vous le voyez avec tous les glycanes, il est presque méconnaissable », explique Amaro, informaticien en chimie biophysique (computationnelle) à l’Université de Californie à San Diego. De nombreux virus ont des glycanes recouvrant leurs protéines externes, les camouflant du système immunitaire humain comme un loup déguisé en mouton. Mais l’année dernière, le groupe du laboratoire d’Amaro et ses collaborateurs ont créé la visualisation la plus détaillée à ce jour de cette couche, basée sur des données structurelles et génétiques et rendues, atome par atome, par un superordinateur. Le 22 mars 2020, il a posté la simulation sur Twitter. En moins d’une heure, un chercheur a demandé dans un commentaire : quelle est cette boucle nue et non enduite qui dépassait du sommet de la protéine ?Pointes dissimulées
Amaro n’en avait aucune idée. Mais dix minutes plus tard, le biologiste structural Jason McLellan de l’Université du Texas à Austin est intervenu : la boucle non revêtue était un domaine de liaison au récepteur (RBD), l’une des trois sections de la pointe qui se lient aux récepteurs des cellules humaines (voir « Un pic’).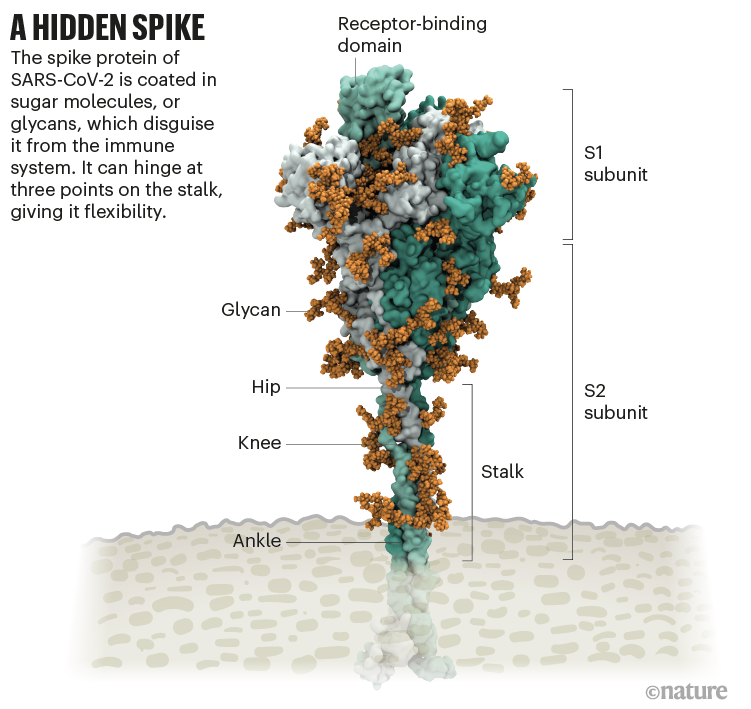
Source : Image structurelle de Lorenzo Casalino, Univ. Californie, San Diego (Réf. 1); Graphique : Nik Spencer/ Nature
Le variant Delta rapide comme l’éclair
Depuis le début de la pandémie de COVID-19, les scientifiques ont développé une compréhension détaillée de la façon dont le SRAS-CoV-2 infecte les cellules. En séparant le processus d’infection, ils espèrent trouver de meilleurs moyens de l’interrompre grâce à des traitements et des vaccins améliorés, et apprendre pourquoi les dernières souches, telles que la variante Delta, sont plus transmissibles. Ce qui a émergé de 19 mois de travail, soutenu par des décennies de recherche sur le coronavirus, est un compte rendu détaillé de la façon dont le SRAS-CoV-2 envahit les cellules humaines (voir « Cycle de vie du coronavirus pandémique »). Les scientifiques ont découvert des adaptations clés qui aident le virus à s’accrocher aux cellules humaines avec une force surprenante, puis à se cacher une fois à l’intérieur. Plus tard, lorsqu’il quitte les cellules, le SARS-CoV-2 exécute une étape de traitement cruciale pour préparer ses particules à infecter encore plus de cellules humaines. Ce sont quelques-uns des outils qui ont permis au virus de se propager si rapidement et de faire des millions de morts. «C’est pourquoi c’est si difficile à contrôler», explique Wendy Barclay, virologue à l’Imperial College de Londres.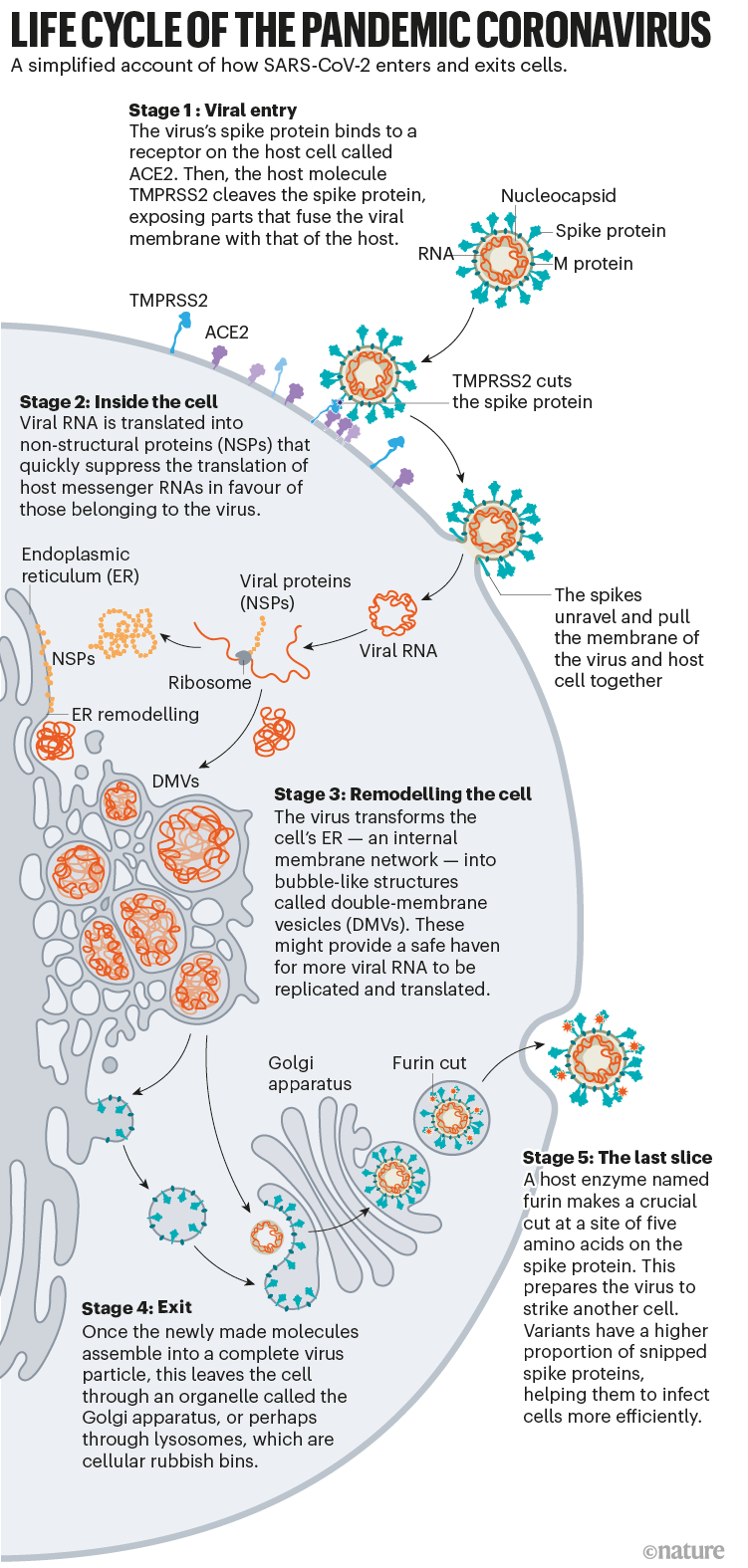
Source : Hui (Ann) Liu, Univ. Utah ; Graphique : Nik Spencer/ Nature
Acéré comme du Barbelé et prêt à surgir
Cela commence par les pointes. Chaque virion (particule virale) du SRAS-CoV-2 a une surface externe parsemée de 24 à 40 protéines de pointe disposées au hasard qui sont la clé de la fusion avec les cellules humaines 2. Pour d’autres types de virus, comme la grippe, les protéines de fusion externes sont relativement rigides. Les pointes du SRAS-CoV-2, cependant, sont extrêmement flexibles et s’articulent en trois points, selon les travaux publiés en août 2020 par le biochimiste Martin Beck de l’Institut Max Planck de biophysique de Francfort, en Allemagne, et ses collègues 3. Cela permet aux pointes de basculer, de se balancer et de tourner, ce qui pourrait leur permettre de scanner plus facilement la surface cellulaire et de permettre à plusieurs pointes de se lier à une cellule humaine. Il n’y a pas de données expérimentales similaires pour d’autres types de coronavirus, mais comme les séquences de protéines de pointe sont hautement conservées au cours de l’évolution, il est juste de supposer que cette caractéristique est partagée, dit Beck.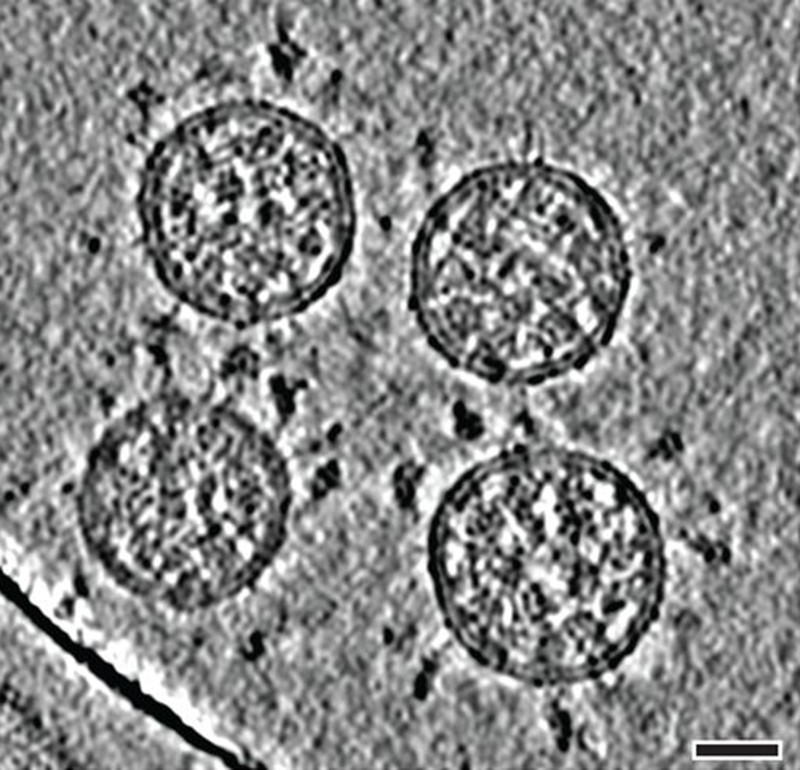
Images de tomographie cryoélectronique des virions du SRAS-CoV-2. (Barre d’échelle : 30 nanomètres.) Crédit : B. Turoňová et al. / Sciences
Variant à mutations multiples
La variante Alpha, par exemple, comprend dix changements dans la séquence des protéines de pointe, ce qui fait que les RBD sont plus susceptibles de rester en position « haute » 6 . « Cela aide le virus à entrer plus facilement dans les cellules », explique Priyamvada Acharya, biologiste structurale au Duke Human Vaccine Institute de Durham, en Caroline du Nord, qui étudie les mutations des pointes. La variante Delta, qui se répand maintenant dans le monde, héberge de multiples mutations dans la sous-unité S1, dont trois dans le RBD qui semblent améliorer la capacité du RBD à se lier à l’ACE2 et à échapper au système immunitaire7 .Entrée restreinte
Une fois que les pointes virales se lient à l’ACE2, d’autres protéines à la surface de la cellule hôte initient un processus qui conduit à la fusion des membranes virales et cellulaires (voir « Entrée virale de près »).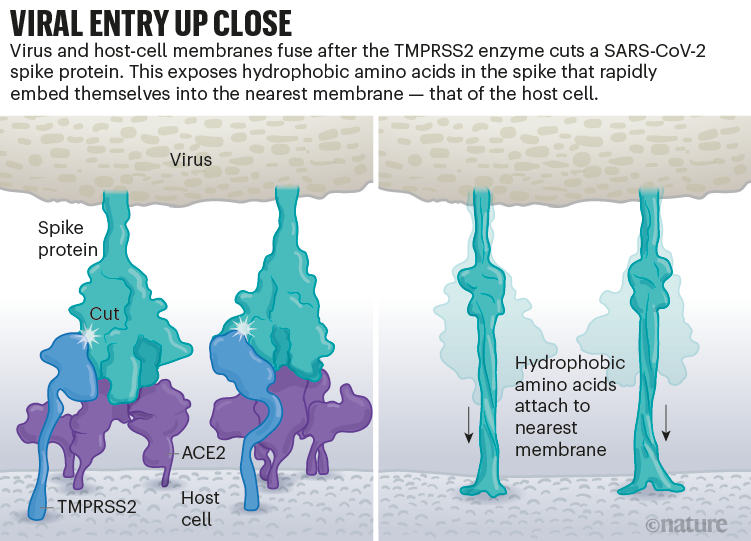
Source : Janet Iwasa, Univ. Utah ; Graphique : Nik Spencer/ Nature
Passé maître dans l’art de la fusion cellulaire
Le SARS-CoV-2 diffère du SARS-CoV car il utilise efficacement TMPRSS2, une enzyme trouvée en grande quantité à l’extérieur des cellules respiratoires. Tout d’abord, TMPRSS2 rompt un site sur la sous-unité S2 du pic 8 . Cette coupe expose une série d’acides aminés hydrophobes qui s’enfouissent rapidement dans la membrane la plus proche, celle de la cellule hôte. Ensuite, la pointe étendue se replie sur elle-même, comme une fermeture éclair, forçant les membranes virales et cellulaires à fusionner.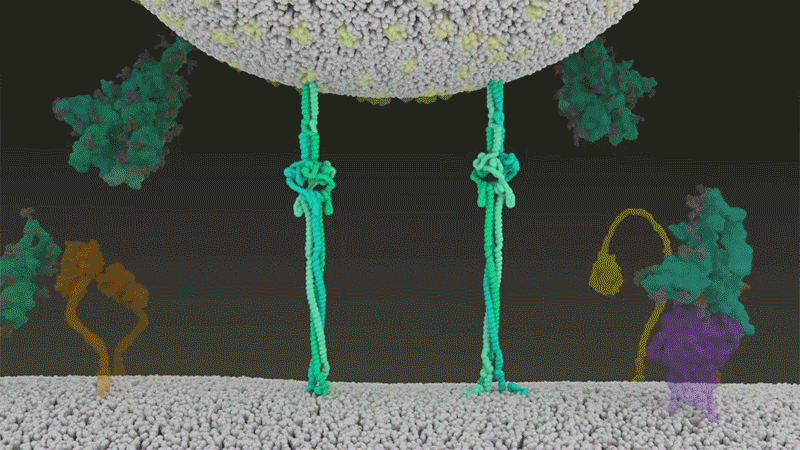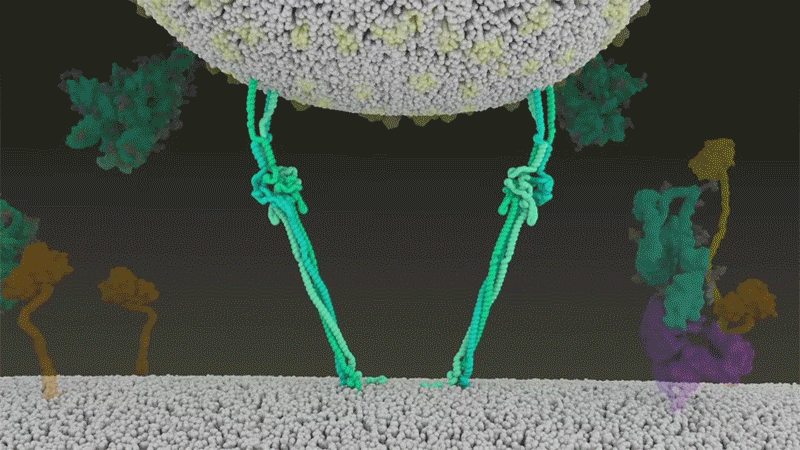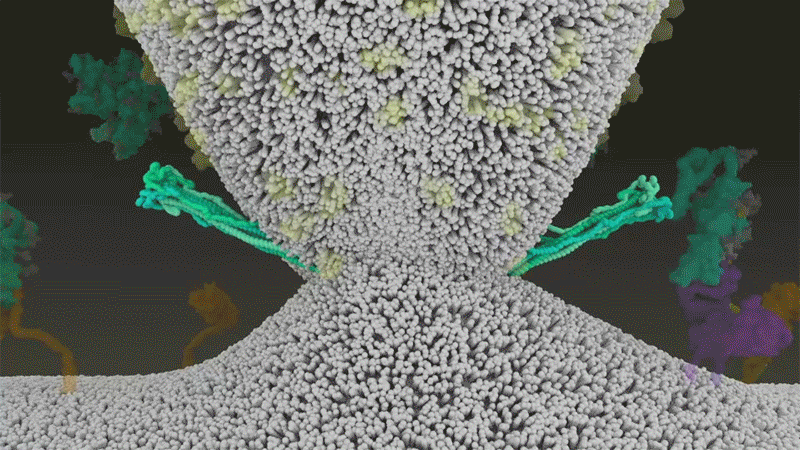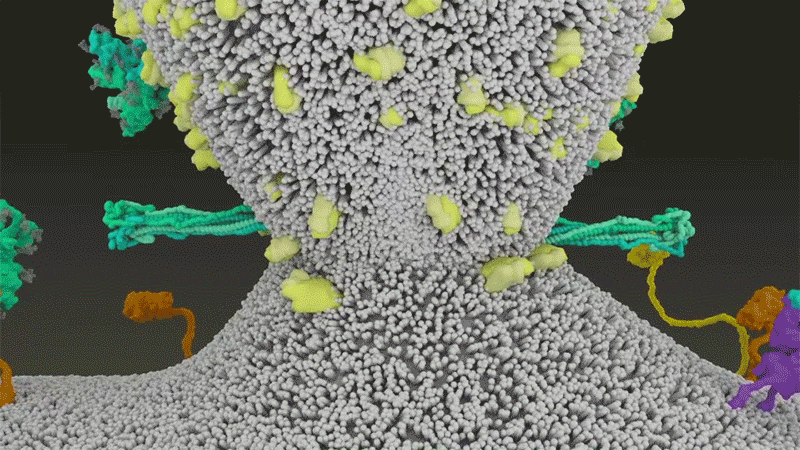
Une animation de la façon dont le SARS-CoV-2 fusionne avec les cellules. Crédit : Janet Iwasa, Université de l’Utah
Pourquoi les adeptes de la Chloroquine se sont mis le doigt dans les endosomes
L’entrée rapide du virus à l’aide de TMPRSS2 explique pourquoi la chloroquine, un médicament contre le paludisme, n’a pas fonctionné dans les essais cliniques en tant que traitement COVID-19, malgré les premières études prometteuses en laboratoire 10 . Ceux-ci se sont avérés avoir utilisé des cellules qui dépendent exclusivement des cathepsines pour l’entrée endosomale. « Lorsque le virus se transmet et se réplique dans les voies respiratoires humaines, il n’utilise pas d’endosomes, donc la chloroquine, qui est un médicament perturbateur endosomal, n’est pas efficace dans la vraie vie », explique Barclay. La découverte indique également que les inhibiteurs de protéase constituent une option thérapeutique prometteuse pour empêcher un virus d’utiliser TMPRSS2, la cathepsine L ou d’autres protéases pour pénétrer dans les cellules hôtes. Un inhibiteur de TMPRSS2, le mésylate de camostat, approuvé au Japon pour traiter la pancréatite, a bloqué l’entrée virale dans les cellules pulmonaires 8, mais le médicament n’a pas amélioré les résultats des patients lors d’un essai clinique initial 11.En quête d’antiviraux
« De mon point de vue, nous devrions avoir des inhibiteurs de protéase tels que de larges antiviraux disponibles pour lutter contre les nouvelles épidémies et prévenir de futures pandémies dès le tout début », déclare Stefan Pöhlmann, directeur de l’unité de biologie des infections au German Primate Center de Göttingen, qui a a dirigé des recherches sur la liaison ACE2 et la voie TMPRSS2.Compétition mortelle
Les prochaines étapes de l’infection sont plus troubles. « Il y a beaucoup plus de boîtes noires une fois que vous êtes à l’intérieur de la cellule », explique la chimiste Janet Iwasa de l’Université de l’Utah à Salt Lake City, qui développe une animation annotée du cycle de vie viral. « Il y a plus d’incertitude et d’hypothèses concurrentes. »Comment le virus réplique des copies de son ARN messager
Une fois que le virus a envoyé son ARN génomique dans la cellule, les ribosomes du cytoplasme traduisent deux sections d’ARN viral en longues chaînes d’acides aminés, qui sont ensuite découpées en 16 protéines, dont beaucoup sont impliquées dans la synthèse de l’ARN. Plus tard, davantage d’ARN sont générés qui codent pour un total de 26 protéines virales connues, y compris les protéines structurelles utilisées pour fabriquer de nouvelles particules virales, telles que la pointe, et d’autres protéines accessoires. De cette façon, le virus commence à produire des copies de son propre ARN messager. Mais il a besoin de la machinerie de la cellule pour traduire ces ARNm en protéines.
Comment une variante endémique du coronavirus émousse nos défenses immunitaires
Le virus cambrioleur déconnecte les alarmes
Deuxièmement, l’infection réduit la traduction globale des protéines dans la cellule de 70 %. Nsp1 est à nouveau le principal coupable, bloquant cette fois physiquement le canal d’entrée des ribosomes, afin que l’ARNm ne puisse pas y pénétrer, selon les travaux de deux équipes de recherche 13 , 14. La petite capacité de traduction qui reste est dédiée aux ARN viraux, explique Stern-Ginossar. Enfin, le virus arrête le système d’alarme de la cellule. Cela se produit de nombreuses manières, mais l’équipe de Stern-Ginossar a identifié un mécanisme clair pour le SRAS-CoV-2 : le virus empêche l’ARNm cellulaire de sortir du noyau, y compris d’envoyer des instructions pour les protéines destinées à alerter le système immunitaire en cas d’infection. Une deuxième équipe a confirmé cette découverte, et a de nouveau pointé du doigt Nsp1 : la protéine semble bloquer les canaux de sortie dans le noyau afin que rien ne puisse s’échapper 15 . Parce que les transcrits génétiques ne peuvent pas sortir du noyau, les cellules infectées ne libèrent pas beaucoup d’interférons – ce sont des protéines de signalisation qui alertent le système immunitaire de la présence d’un virus. Le SARS-Cov-2 est particulièrement efficace pour arrêter ce système d’alarme : par rapport à d’autres virus respiratoires, y compris le SARS-CoV et le virus respiratoire syncytial, l’infection par le SARS-CoV-2 induit des niveaux d’interférons significativement plus faibles 16 . Et en juin dernier, des chercheurs ont signalé des mutations dans la variante Alpha qui semblent lui permettre de maîtriser encore plus efficacement la production d’interféron 17 .Le virus déjoue les capteurs immunitaires
« Il est clair que le SRAS-CoV-2 est un virus très rapide qui a une capacité unique à empêcher notre système immunitaire de reconnaître et de combattre l’infection dans les premiers stades », explique Stern-Ginossar. Au moment où le système immunitaire réalise qu’il y a un virus, il y en a tellement que les protéines de réponse immunitaire inondent parfois la circulation sanguine à un rythme plus rapide que la normale, ce qui peut causer des dommages. Les médecins ont constaté au début de la pandémie que certaines personnes atteintes de COVID-19 qui tombent très malades sont lésées par une réponse immunitaire hyperactive au SRAS-CoV-2, ainsi que par le virus lui-même. Certains traitements éprouvés agissent en atténuant cette réponse immunitaire.Poste de régénération (cure de jouvence)
Une fois que le virus a pris en charge la traduction de l’hôte, il commence une cure de jouvence, remodelant largement l’intérieur et l’extérieur de la cellule selon ses besoins. Premièrement, certaines des protéines de pointe virales nouvellement fabriquées se déplacent à la surface de la cellule et sortent de la membrane de la cellule hôte. Là, ils activent un canal ionique calcium hôte, qui expulse un revêtement gras à l’extérieur de la cellule – le même revêtement que l’on trouve sur les cellules qui fusionnent naturellement, telles que les cellules musculaires. À ce stade, la cellule infectée fusionne avec les cellules voisines exprimant l’ACE2, se développant en cellules respiratoires individuelles massives contenant jusqu’à 20 noyaux.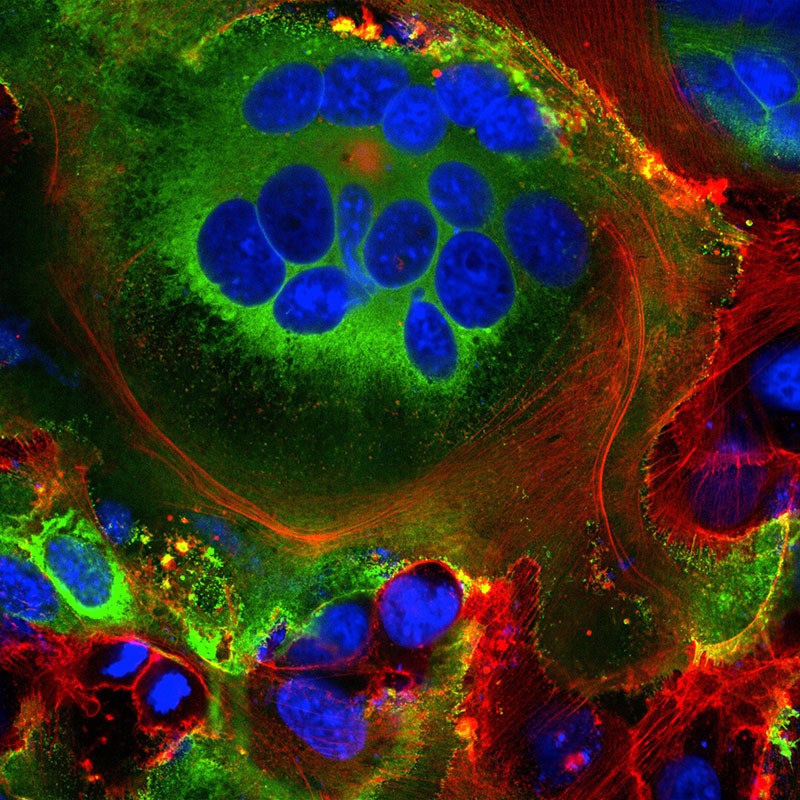
Structures cellulaires fusionnées (syncytia) observées dans les cellules exprimant la protéine de pointe SARS-CoV-2 (vert). Les noyaux sont en bleu et le squelette cellulaire est en rouge. Crédit : Mauro Giacca
Une affection de longue durée
Ces structures fusionnées, appelées syncytia, sont induites par des infections virales telles que le VIH et le virus de l’herpès simplex, mais pas par le virus du SRAS, explique le biologiste moléculaire Mauro Giacca du King’s College de Londres, qui a dirigé l’équipe qui a publié les résultats le 18 avril . Il émet l’hypothèse que la formation de syncytia permet aux cellules infectées de se développer pendant de longues périodes, produisant de plus en plus de virions. « Ce n’est pas un virus hit-and-run » (qui frappe et s’enfuit), dit-il. « Ça persiste. » Une deuxième équipe, dirigée par le chercheur Qiang Sun de l’Académie chinoise des sciences médicales à Pékin, a découvert que certaines cellules infectées par le COVID-19 forment même des syncytia avec des lymphocytes – l’une des propres cellules immunitaires du corps 19. Il s’agit d’un mécanisme connu d’évasion immunitaire par les cellules tumorales, mais pas par les virus. Cela suggère que les cellules infectées évitent la détection immunitaire en s’accrochant simplement et en fusionnant avec les éclaireurs immunitaires à proximité. À l’intérieur de la cellule, encore plus de changements se produisent. Comme d’autres coronavirus, le SRAS-CoV-2 transforme le long et mince réticulum endoplasmique (RE), un réseau de membranes plates impliquées dans la synthèse et le transport des protéines, en sphères à double membrane, comme si le RE soufflait des bulles. Ces vésicules à double membrane (DMV) pourraient fournir un endroit sûr pour la réplication et la traduction de l’ARN viral, le protégeant des capteurs immunitaires innés dans la cellule, mais cette hypothèse est toujours à l’étude.Repérer des cibles médicamenteuses
Les protéines impliquées dans la fabrication des DMV pourraient être de bonnes cibles médicamenteuses, car elles semblent être nécessaires à la réplication virale. Par exemple, une protéine hôte, TMEM41B, est nécessaire pour mobiliser le cholestérol et d’autres lipides afin d’élargir les membranes du RE, de façon à ce que toutes les parties du virus s’intègrent à l’intérieur 20 . « Lorsque vous retirez TMEM41B, cela a un impact majeur sur l’infection », explique Vineet Menachery, chercheur sur les coronavirus à la branche médicale de l’Université du Texas à Galveston, qui a participé à la recherche. La protéine transmembranaire du coronavirus Nsp3 pourrait également être une cible : elle crée un pore en forme de couronne dans les parois des DMV pour faire sortir l’ARN viral nouvellement fabriqué 21 . La plupart des virus qui ont une enveloppe externe, connue sous le nom d’enveloppe, forment cette caractéristique en s’assemblant directement au bord de la cellule, cooptant une partie de la propre membrane plasmique de la cellule lors de sa sortie. Mais les protéines de coronavirus nouvellement fabriquées empruntent un chemin différent.Des virions crachés comme d’une poulpe
Pendant des années, des preuves ont suggéré que le coronavirus est transporté hors de la cellule par le complexe de Golgi, un organite qui fonctionne comme un bureau de poste, emballant des molécules dans des membranes et les envoyant vers d’autres parties de la cellule. Là, le virus forme une enveloppe lipidique à partir de la membrane du complexe de Golgi ; Les virions nouvellement formés sont ensuite transportés à l’intérieur des vésicules de Golgi jusqu’à la surface cellulaire, où ils sont crachés hors de la cellule, explique la virologue et biologiste cellulaire Carolyn Machamer de l’Université Johns Hopkins de Baltimore, Maryland, qui a étudié les coronavirus pendant 30 ans. Mais en décembre, la biologiste cellulaire Nihal Altan-Bonnet du US National Heart, Lung and Blood Institute à Bethesda, Maryland, et ses collègues ont signalé qu’ils avaient détecté des formes de coronavirus quittant la cellule à travers des lysosomes – des poubelles cellulaires pleines d’enzymes qui se décomposent en parties cellulaires 22 . Le blocage de la voie sécrétoire basée sur Golgi n’a pas semblé affecter la quantité de virus infectieux libéré, explique Altan-Bonnet. Les preuves de son équipe 22 suggèrent que les protéines virales forment une enveloppe en bourgeonnant dans le RE, puis prennent le relais des lysosomes pour sortir de la cellule. Les chercheurs testent actuellement des inhibiteurs qui bloquent le processus de sortie lysosomale en tant que candidats antiviraux potentiels.Des phases encore inexpliquées
Laisser une cellule à travers le Golgi ou les lysosomes est lent et inefficace par rapport au bourgeonnement d’une membrane plasmique, de sorte que les scientifiques ne savent pas pourquoi le SRAS-CoV-2 le fait. Machamer soupçonne que la composition lipidique d’une enveloppe dérivée de Golgi ou de lysosome est en quelque sorte plus bénéfique pour le virus que celle de la membrane plasmique. « Si nous comprenions un peu mieux cette partie, il y aurait de grandes opportunités pour de nouvelles thérapies antivirales », dit-elle.Dernière tranche
À la sortie de la cellule, un événement de plus fait de ce virus un mastodonte infectieux : une coupe rapide sur un site de cinq acides aminés prépare le virus à frapper sa prochaine cible. Là où d’autres coronavirus ont un seul acide aminé arginine à la jonction des sous-unités S1 et S2 de la pointe, le SRAS-CoV-2 a une ligne de cinq acides aminés : proline, arginine, arginine, alanine et à nouveau arginine. « Parce que le site était inhabituel, nous nous sommes concentrés sur lui, et il s’est avéré que, oui, le site est essentiel pour l’invasion des cellules pulmonaires », explique Pöhlmann. En mai 2020, lui et ses collègues ont signalé qu’une protéine de la cellule hôte appelée furine reconnaît et coupe cette chaîne d’acides aminés – et la coupure est « essentielle » pour que le virus pénètre efficacement dans les cellules pulmonaires humaines 23 . Ce n’est pas la première fois que des chercheurs identifient un site de clivage de la furine sur un virus ; les virus hautement pathogènes de la grippe aviaire en sont également atteints, dit Barclay. Lorsqu’un collègue a envoyé à Barclay une souche de SRAS-CoV-2 en culture qui avait spontanément perdu le site de clivage de la furine, son équipe a découvert que les furets infectés par cette souche libéraient des particules virales en quantités inférieures à celles infectées par la souche pandémique, et ont fait en sorte qu’ils ne transmettent pas l’infection aux animaux à proximité 9 . En même temps que l’équipe de Barclay rapportait ses résultats dans une préimpression de septembre 2020, une étude aux Pays-Bas a également révélé que le coronavirus avec un site de clivage de la furine intact pénètre plus rapidement dans les cellules des voies respiratoires humaines que celles qui n’en ont pas 24 .Le rôle de la furine
La furine est soupçonnée de couper le site à un moment donné pendant l’assemblage du virion, ou juste avant la libération. Le timing pourrait expliquer pourquoi le virus sort par le Golgi ou les lysosomes, explique Tom Gallagher, virologue à l’Université Loyola de Chicago dans l’Illinois. « Le virus, une fois assemblé, se déplace dans un organite où il peut être baigné en présence de la protéase furine. » En coupant la liaison entre les sous-unités S1 et S2, la coupe de furine relâche les protéines de pointe du virion de sorte que lors de l’entrée dans la cellule, elles répondent à une deuxième coupe par TMPRSS2, qui expose la zone hydrophobe qui s’enfouit rapidement dans une membrane de cellule hôte, dit Gallagher. Si les pointes ne sont pas pré-coupées par la furine – et elles ne le sont pas toujours – elles contournent TMPRSS2 et pénètrent par la voie endosomale plus lente, voire pas du tout.
La course aux médicaments antiviraux pour vaincre le COVID – et la prochaine pandémie
Avec le Delta, plus de 75% de protéines de pointes amorcées
Deux variantes de coronavirus, Alpha et Delta, ont altéré les sites de clivage de la furine. Dans la variante Alpha, l’acide aminé proline initial est changé en histidine (P681H) ; dans la variante Delta, il est remplacé par une arginine (P681R). Les deux changements rendent la séquence moins acide, et plus la chaîne d’acides aminés est basique, plus la furine la reconnaît et la coupe efficacement, explique Barclay. « Nous émettrons l’hypothèse que c’est le virus qui se transmet encore mieux. » Plus de coupures de furine signifient plus de protéines de pointe amorcées pour entrer dans les cellules humaines. Dans le SRAS-CoV, moins de 10 % des protéines de pointe sont amorcées, explique Menachery, dont le groupe de laboratoire a quantifié les protéines de pointe amorcées mais n’a pas encore publié ce travail. Dans le SRAS-CoV-2, ce pourcentage monte à 50 %. Dans la variante Alpha, c’est plus de 50 %. Dans la variante Delta hautement transmissible, le groupe a découvert que plus de 75 % des pointes sont amorcées pour infecter une cellule humaine.Inconnues et choses mieux connues
La communauté scientifique effleure encore la surface de sa compréhension du SARS-CoV-2. Les principales inconnues incluent le nombre de récepteurs ACE2 nécessaires pour se lier à chaque protéine de pointe ; quand exactement le site S2 est clivé par TMPRSS2 ; et le nombre de pointes nécessaires à la fusion virus-membrane cellulaire, dit McLellan – et c’est juste pour l’entrée. En avril 2020, une équipe de l’Université de Californie à San Francisco a identifié au moins 332 interactions entre le SRAS-CoV-2 et les protéines humaines 25 .Propagation plus rapides ou plus de dommages causés individuellement?
Il n’est pas facile de suivre le rythme de la mutation rapide du virus. Jusqu’à présent, la plupart des mutations sont associées à l’efficacité de la propagation du virus, et non à l‘ampleur des dommages causés par le virus à l’hôte, conviennent les experts. Ce mois-ci, une étude a rapporté que la variante Delta s’est développée plus rapidement et à des niveaux plus élevés dans les poumons et la gorge des personnes que les versions antérieures du virus 26 . Mais on ne sait pas encore avec certitude comment les mutations de Delta ont suralimenté le variant de cette manière, explique Stern-Ginossar. « C’est quelque chose que de nombreux laboratoires essaient de comprendre. »Les références
- 1.
Casalino, L. et al. ACS Cent. Sci. 6 , 1722-1734 (2020).
- 2.
Ke, Z. et al. Nature 588 , 498-502 (2020).
- 3.
Turoňová, B. et al. Sciences 370 , 203-208 (2020).
- 4.
Nguyen, HL et al. J. Phys. Chem. B 124 , 7336-7347 (2020).
- 5.
Shang, J. et al. Nature 581 , 221-224 (2020).
- 6.
Gobeil, SM-C. et al. Sciences https://doi.org/10.1126/science.abi6226 (2021).
- 7.
Khateeb, J., Li, Y. & Zhang, H. Crit. Soins 25 , 244 (2021).
- 8.
Hoffmann, M. et al. Cellule 181 , 271-280 (2020).
- 9.
Peacock, TP et al. Microbiol naturel. 6 , 899-909 (2021).
- dix.
Wang, M. et al. Rés. 30 , 269-271 (2020).
- 11.
Gunst, JD et al. EClinicalMedicine 35 , 100894 (2021).
- 12.
Finkel, Y. et al. Nature 594 , 240-245 (2021).
- 13.
Schubert, K. et al. Structure de la nature. Mol. Biol. 27 , 959-966 (2020).
- 14.
Thoms, M. et al. Sciences 369 , 1249-1255 (2020).
- 15.
Zhang, K. et al. Sci. Av. 7 , eabe7386 (2021).
- 16.
Blanco-Melo, D. et al. Cellule 181 , 1036-1045 (2020).
- 17.
Thorne, LG et al. Préimpression sur bioRxiv https://doi.org/10.1101/2021.06.06.446826 (2021).
- 18.
Braga, L. et al. Nature 594 , 88-93 (2021).
- 19.
Zhang, Z. et al. La mort cellulaire diffère . https://doi.org/10.1038/s41418-021-00782-3 (2021).
- 20.
Trimarco, JD et al. Pathog PLoS. 17 , e1009599 (2021).
- 21.
Wolff, G. et al. Sciences 369 , 1395-1398 (2020).
- 22.
Ghosh, S. et al. Cellule 183 , 1520-1535 (2020).
- 23.
Hoffmann, M., Kleine-Weber, H. & Pöhlmann, S. Mol. Cellule 78 , 779-784 (2020).
- 24.
Mykytyn, AZ et al. eLife 10 , e64508 (2021).
- 25.
Gordon, DE et al. Nature 583 , 459-468 (2020).
- 26.
Li, B. et al. Préimpression sur medRxiv https://doi.org/10.1101/2021.07.07.21260122 (2021).